


La Fondation Stargardt soutient et co-finance chaque année un projet de recherche dont l’objectif d’étude est lié à l’amélioration des mécanismes de la vision impliquée dans les maculopathies dont fait partie la maladie de Stargardt. Le but à terme de l’ensemble de ces recherches est d’apporter des solutions dans le traitement et/ou la prévention des maculopathies.
Un nouveau projet avec l’Université de Strasbourg en 2024
Pour que les projets réalisés en recherche médicale sur la maladie de Stargardt gagnent en efficacité, il est essentiel de trouver des modèles d’expérimentation animales qui soient plus proches de celui de l’homme.
La Fondation STARGARDT finance, cette année, un nouveau projet de recherche en collaboration avec le CNRS et l’Institut des Neurosciences Cellulaires et Intégratives (INCI) de l’Université de Strasbourg.

L’équipe de recherche encadré par le professeur David Hicks est parvenue à mettre au point un protocole d’injection intraoculaire chez une espèce de rongeurs (« Psammomys »), proche de la souris, mais avec des yeux actifs le jour.
Cela dit, pour atteindre les essais précliniques, le modèle doit encore passer, pendant environ 3 mois, des contrôles scientifiques. La Fondation a décidé de soutenir le projet le long de cette période de tests.
Dans l’article ci-dessous, le Pr. Hicks nous décrit son projet avec plus de détails.
Qui est le Pr. David HICKS ?

David Hicks est directeur de recherches de l’INSERM à l’Institut des neurosciences cellulaires et intégratives (INCI) à Strasbourg. Il s’est formé à la zoologie à l’université de Bristol, Royaume-Uni, et a obtenu son doctorat en neurobiologie à l’université de Londres en 1981.
Il a commencé sa carrière de chercheur en Amérique du Nord, d’abord dans l’université de la Colombie-Britannique à Vancouver (Canada) puis, dans le Laboratoire de neurobiologie dirigé par le Professeur Torsten Wiesel, prix Nobel de physiologie et médecine 1981, à l’université Rockefeller de New York.
Depuis ses études postdoctorales, le Pr. Hicks s’intéresse aux mécanismes cellulaires et moléculaires impliqués dans la différenciation, la fonction et la survie de la rétine.
Chercheur de l’Inserm depuis 1988, il est actuellement co-directeur du groupe « Rythmes, vie et mort dans la rétine » à l’INCI, où il travaille sur le rôle de l’horloge circadienne dans la physiopathologie de la rétine et le rôle central des photorécepteurs de type cône dans la vision.
Le Pr. Hicks s’adresse aux membres de la Fondation
« La maladie de Stargardt (MS) constitue une maladie génétique de l’oeil touchant principalement les jeunes, menant à une malvoyance parfois profonde et orpheline de traitement à ce jour. Le gène » Abca4 » -dont les mutations sont responsables de la maladie- est identifié depuis longue date.
Ceci a permis de créer rapidement des modèles animaux où le gène est inactivé et d’examiner les conséquences sur la survie et la fonction de la rétine – nous explique le Professeur Hicks.
Toutefois, jusqu’à présent, la recherche sur la maladie de Stargardt utilisait principalement des rongeurs de nuit dans ses essais. Ces animaux sont des souris » abca4 knockout » qui, même s’ils partagent certaines caractéristiques de la maladie de Stargardt humaine (accumulation de dépôts lipidiques, ou « lipofuscine »), globalement, ils ne récapitulent pas ou peu la vraie maladie, à savoir, ils ne subissent pas de dégénérescence tissulaire.
Dans ce sens, l’obtention de modèles animaux pertinents est capitale pour approfondir nos connaissances biomédicales et fournir un moyen de tester les remèdes possibles. Ce manque a longtemps freiné les recherches.
Par exemple, à ce jour, la recherche n’est pas en mesure d’expliquer pourquoi la maladie de Stargardt atteint majoritairement les photorécepteurs de type cône alors que le gène abca4 est présent dans les cônes et les bâtonnets. »
FS : Mais…quelle est la différence entre les cônes et les bâtonnets ?
« Les cônes se trouvent très concentrés au niveau de la macula, et ils sont essentiels pour la vision colorée et détaillée chez l’Humain. En revanche les bâtonnets permettent de voir en conditions de faible luminosité, par exemple, le soir. »
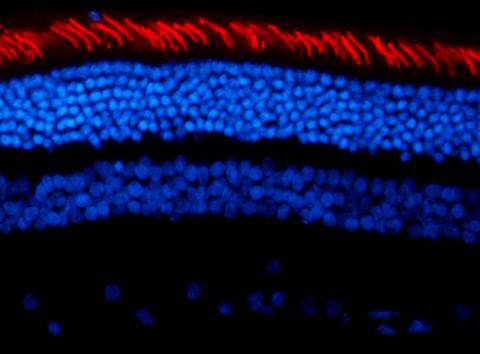
« La maladie de Stargardt est certes une maladie d’une sévérité variable, mais elle concerne dans la majorité des cas, une atteinte de la macula. Par exemple, parfois il est question d’une lésion uniquement et…encore plus étrange, il y a des formes de la maladie où les bâtonnets fonctionnent de façon normale alors que les cônes sont endommagés.» nous indique-t-il.
Prenant note de ces caractéristiques, l’équipe du Professeur Hicks a émis l’hypothèse suivante : les souris sont mal-adaptées pour servir comme modèle pour la maladie de Stargardt.
« La raison est simple, elles n’ont que très peu de cônes (<3%). Les souris sont des animaux naturellement nocturnes, ce qui veut dire qu’elles n’ont pas besoin d’une vision très précise.
C’est de ce constat que naît l’intérêt de modéliser les pathologies rétiniennes avec des rongeurs diurnes, actifs le jour, qui possèdent un pourcentage élevé de cônes (~33%) similaire à la rétine centrale humaine.
L’INCI est l’une des très rares institutions dans le monde à héberger des colonies de ces rongeurs. Lors de travaux antérieurs, ces animaux ont servi pour mettre en évidence certaines différences entre les cônes et les bâtonnets qui pourraient expliquer la vulnérabilité accrue des premiers. Il fallait prouver que les cônes soient moins résistants que les bâtonnets dans le contexte de la maladie de Stargardt.
Pour ce faire, nous avons endormi le » gène abca4 » par injection intra-oculaire de virus portant une construction » CRISPR-Cas9 » spécifique. Cette nouvelle technologie qui permet de « bricoler » assez aisément les gènes pour altérer voire inhiber totalement leur fonction.
La recherche n’était pas facile pour autant, et nous avons dû faire face à de nombreux obstacles scientifiques et techniques. Mais après quatre ans de travail intense, nous sommes finalement parvenus à mettre au point un protocole d’injection intraoculaire chez ces jeunes « Psams ». Comparé aux animaux témoins (injectés avec un virus portant une sonde neutre sans activité), l’examen du fond d’oeil a révélé une dégénérescence profonde des photorécepteurs.»

« L’analyse fonctionnelle des animaux par « électrorétinographie » -technique permettant de mesurer la sensibilité visuelle- suivie par des examens postmortem en histologie, immunochimie et biologie moléculaire, ont prouvé que la dégénérescence causée par la maladie est essentiellement due à la perte des cônes.
L’ensemble des résultats a permis d’affirmer que ce modèle est bien le premier à récapituler assez fidèlement la maladie de Stargardt humaine– disparition des cônes, perte de sensibilité visuelle, accumulation de la lipofuscine…) et devrait ouvrir des axes forts intéressants en recherche scientifiques et thérapeutiques. »
—————–
Le mot de la Fondation
« Ce modèle » Psammomys » de rongeur découvert par le professeur Hicks aura pour objectif d’étudier avec beaucoup plus de facilité la vulnérabilité des photorécepteurs au fur et à mesure que l’affection du gène Abca4 – responsable de la maladie- évolue.
L’utilisation de ce rongeur par les laboratoires pharmaceutiques ou par les sociétés de biotechnologies souhaitant développer des médicaments pour soigner la maladie de Stargardt, leur permettra d’apporter aux agences règlementaires des preuves d’efficacité plus solides pour permettre le passage vers l’essai clinique sur l’homme. »
Les programmes de recherche financés par la Fondation en 2022
Projets développés par l’Inserm Montpellier
Rappel sur le projet de médicament de Philippe BRABET : Les travaux portent sur l’étude d’une nouvelle molécule pharmacologique, IP-DHA dont le mode d’action repose sur la diminution de l’accumulation de métabolites toxiques (pharmacothérapie). Cette recherche est réalisée par une équipe multicentrique dont les Dr Philippe BRABET et Vasiliki KALATZIS de l’INSERM Montpellier.
Introduction : Depuis 2021, La Fondation finance avec l’IRRP (Information Recherche Rétinite Pigmentaire) les travaux initiés il y a près de 10 ans par le Dr Philippe BRABET. Depuis 2021, Philippe BRABET travaille avec Vasiliki KALATZIS pour mieux comprendre le développement « génétique » de la maladie de Stargardt. Pour ce faire, depuis 2021 les chercheurs utilisent des organoïdes (rétine refaite à partir de la peau de malade de Stargardt) pour tenter de mieux comprendre pourquoi l’évolution d’une rétine de Stargardt ne va pas toujours dans la bonne direction !
Les subventions de la Fondation Stargardt ont permis à l’équipe du Dr. Philippe Brabet de réaliser des études pharmacologiques et de tester de nouvelles molécules à visée thérapeutiques en collaboration étroite avec deux équipes, celle de Thierry Durand et Céline Crauste à l’IBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, et de Sylvie Bégu à l’ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier.

Le Dr.BRABET et son assistant, Gregor DUBOIS, à l’Inserm Montpellier
En ce moment, leur projet de recherche est axé sur trois aspects principaux. Le premier aspect est focalisé sur les mécanismes d’action de la molécule. Il est en effet important de savoir comment cette molécule, l’IP-DHA, est capable de protéger la rétine. L’IP-DHA est composée de 3 éléments : un polyphénol (le phloroglucinol), un acide gras oméga 3 (le DHA) et un isopropyl. Après avoir réalisé des tests, les chercheurs se sont rendu compte que paradoxalement, des modifications chimiques visant à stabiliser cette molécule ont été contre-productif. En effet, les trois molécules constituant l’IP-DHA doivent pouvoir se séparer à l’intérieur de la cellule pour garantir son efficacité. Ils se sont aperçus que toutes les parties de la molécule sont nécessaires mais avec des fonctions différentes. Les acides gras permettent à la molécule de rentrer dans la cellule (le DHA) alors que le phloroglucinol neutralise les composés toxiques accumulés dans les cellules de la rétine (Cia D et al., JCMM 2016 ; Taveau et al., EMM, 2020).
Ils ont aussi réussi à localiser la molécule à l’intérieur même de la cellule, et ainsi pu observer qu’elle se retrouve majoritairement là où s’accumule les composés toxiques.
Enfin, un autre mode d’action indirect de l’IP-DHA est de réguler certains gènes importants dans les voies de régulation détoxifiantes.
Le deuxième aspect, pharmacologique, s’est intéressé à développer un mode d’administration orale de la molécule IP-DHA. Dans la première phase de développement, cette molécule s’est montrée efficace lorsqu’elle est administrée à des souris en injection intraveineuse. Afin de faciliter son futur développement à l’Homme, il était important de trouver des voies d’administration moins contraignantes et tout aussi efficace. Cette équipe a donc travaillé sur des voies d’administration orale en utilisant différentes techniques pour permettre une bonne absorption jusqu’au dans la circulation générale (M. Vincent et al., pharmaceutics, 2022).

L’équipe du Dr.BRABET au laboratoire de l’Inserm Montpellier
Enfin, le troisième aspect étudié fut le développement de cellules rétiniennes à partir de prélèvement de morceau de peau. Il est en effet impossible de prélever directement de la rétine chez les patients, c’est pourquoi les médecins prélèvent une petite partie de la peau, dont les cellules sont ensuite reprogrammées en laboratoire en cellules souches pluripotentes (CSPi pour Cellules Souches Pluripotentes Induites en français). Toujours en laboratoire, ces cellules sont ensuite transformées en cellules de la rétine. Elles portent ainsi les mêmes caractéristiques génétiques que les patients dont elles sont issues. Ce travail sur des échantillons humains, toujours en cours, complète les travaux obtenus sur la souris.
L’équipe du Dr Brabet assisté par Grégor Dubois, a produit des cellules de la rétine à partir d’échantillons d’une patiente suivie au centre de référence des maladies rares de Montpellier (Maolya). Ces travaux sont financés actuellement par la fondation Stargardt avec pour objectif principal d’étudier les conséquences liées à un défaut dans le gène ABCA4, responsable de la maladie de Stargardt, à la fois dans les photorécepteurs grâce à des organoïdes rétiniens (structure multicellulaire tridimensionnelle qui reproduit in vitro la micro-anatomie d’un organe) et dans l’épithélium pigmentaires rétinien(EPR).
Projet développé en collaboration avec le CERTO, le CNRS et VARIANT
Rappel : Au fil des décennies, de nombreuses mutations touchant un grand nombre de gènes ont été identifiées comme responsables de la dégénérescence des photorécepteurs. Malgré les énormes progrès de la recherche au cours des dernières années, ces maladies entraînent toujours une cécité légale en raison du nombre limité d’options thérapeutiques. Trouver une thérapie applicable au plus grand nombre de patients et au plus grand nombre de maladies oculaires reste un enjeu majeur. Le succès récent de la thérapie dite de « remplacement génique » chez les patients atteints d’amaurose congénitale de Leber (ACL) causée par des mutations du gène RPE65 a posé les bases pour étendre cette approche à d’autres dystrophies rétiniennes. Dans ce contexte, les travaux antérieurs de Jerome Roger du CERTO suggèrent que les mutations dominantes liées à CRX, un facteur de transcription essentiel au développement et à la maturation des photorécepteurs, représentent des candidats pertinents pour le remplacement génique. Le domaine d’application thérapeutique est large et pourrait bénéficier à de nombreux patients car plus de 50 mutations du gène CRX sont responsables des dystrophies à cône-bâtonnet (CORD), de l’ACL et de la rétinite pigmentaire (RP).
Le CERTO et le CNRS en collaboration avec la société de biotechnologie VARIANT, ont ainsi développé au cours des 4 dernières années un projet visant à développer un produit de thérapie génique de type AAV qui permettrait de traiter un large spectre de rétinopathies associées à CRX.


Les résultats obtenus à ce jour sont très encourageants sur des modèles d’ACL et de dystrophies des cônes. Fin 2020, le produit a même démontré son efficacité sur une pathologie non liée à CRX. L’équipe est donc maintenant en2022 de tester d’autres modèles de souris, atteintes d’autres pathologies, pour voir si l’efficacité de notre produit est à nouveau confirmé. Si c’était lecas, d’autres maculopathies pourraient bénéficier de ce produit et peut être un groupe pharmaceutique s’engagerait dans un essai clinique sur l’homme ! Nous en saurons plus en début d’année prochaine, soit vers février 2023.

Jerôme ROGER, PhD …
« Le CERTO et VARIANT tiennent à remercier Retina France, la fondation Retina et la Fondation Stargardt pour leur soutien inconditionnel à ce projet préclinique ». Jérome ROGER, PhD
Projet de recherche subventionné en 2020, Dr Philippe Brabet et Vasiliki Kalatzis (Inserm Montpellier)
La Fondation soutient avec l’IRPP en 2020 l’étude d’une nouvelle molécule pharmacologique, IP-DHA, dont le mode d’action repose sur la diminution de l’accumulation de métabolites toxiques (pharmacothérapie).
Cette recherche a été réalisée par une équipe multicentrique dont les Dr Philippe Brabet et Vasiliki Kalatzis de l’Inserm Montpellier.
La lumière environnementale a des effets délétères sur la rétine externe dans les rétinopathies humaines, telles que celles liées à ABCA4 soit la maladie de Stargardt et la dégénérescence maculaire sèche liée à l’âge. Ces effets impliquent un stress carbonyle et oxydant, qui contribuent à la mort des cellules rétiniennes et à la perte de vision. Ici, l’étude a eu pour objectif de tester, sur une souris modèle de la maladie de Stargardt, l’efficacité protectrice d’un nouvel acide gras oméga-3. Il s’agit d’un acide polyinsaturé dérivé de lipophénol, l’isopropyl-phloroglucinol-DHA, appelé IP-DHA. Cette molécule est obtenue à partir d’une molécule naturelle extraite d’algues, le phloroglucinol. Celle-ci possède des propriétés reconnues anti-oxydante et antispasmodique.
Les analyses anatomiques et fonctionnelles ont démontré qu’une seule injection intraveineuse de l’IP-DHA, permettait une diminution, proportionnelle à la dose, de la dégénérescence des photorécepteurs induite par la lumière et une sensibilité visuelle préservée. Cet effet protecteur a persisté pendant 3 mois.
IP-DHA n’a pas affecté le cinétique du cycle visuel in vivo ou ni l’activité de l’isomérase RPE65 in vitro. De plus, l’administration d’IP-DHA par voie orale a montré une protection significative des photorécepteurs contre les dommages aigus de la lumière. En conclusion, les tests à court terme, après administration d’une dose unique et selon l’exposition à la lumière, montrent l’IP-DHA comme agent thérapeutique prometteur pour la prévention de la dégénérescence rétinienne.
Pour l’instant, aucun essai chez l’homme n’a été encore réalisé.
Publication Source : Taveau, N., Cubizolle, A., Guillou, L. et al. Preclinical pharmacology of a lipophenol in a mouse model of light-induced retinopathy. Exp Mol Med 52, 1090–1101 (2020). https://doi.org/10.1038/s12276-020-0460-7
Projet de recherche subventionné en 2020, INM (Institut de Neurosciences de Montpellier), INSERM
Le projet porte sur le développement et l’utilisation de modèles cellulaires CRX rétiniens humains pour le développement de nouvelles thérapies.
Également engagé à Montpellier au sein de l’INM (Institut de Neurosciences de Montpellier), INSERM, ce nouveau projet co-financé par la Fondation Stargardt et la société de biotechnologie VARIANT vise à mieux comprendre le développement de la rétine chez les patients atteints d’une maculopathie, similaire à Stargardt, causée par une mutation sur le gène CRX.
Pour ce faire, des extraits de peau sont prélevés sur des malades. Ces fibroblastes sont reprogrammés en cellules souches pluripotentes induites (iPSC), cellules pouvant être différenciées en cellules spécifiques, ici des cellules rétiniennes. Ensuite, l’INM travaille sur les différences phénotypiques (ou Comment grandit la rétine? Quelle protéine lui manque-t-il?, Pourquoi va apparaître la maladie ?)
Projet de recherche subventionné en 2019, Dr Claire-Marie DHAENENS (Lille)
Le projet a pour but de « Mieux comprendre les mutations à l’origine de la maladie de Stargardt pour mieux diagnostiquer dès 2020 et peut-être, traiter certaines formes de la maladie ».
En 2019, la Fondation Stargardt soutient le projet de recherche du Dr Claire-Marie DHAENENS (UF Génopathies, Centre de Biologie Pathologie Génétique, CHRU Lille), en mission dans le laboratoire du Pr Frans CREMERS de l’université Radboud à Nimègue aux Pays-Bas. Le projet porte sur la recherche d’un certain type de mutations du gène ABCA4, appelées mutations introniques profondes : « Recherche et caractérisation des mutations introniques profondes dans une cohorte de 224 patients atteints de la maladie de Stargardt ».
Pour rappel, la maladie de Stargardt (STGD1) est due à la présence de deux mutations bi-alléliques dans le gène ABCA4, qui est le gène le plus fréquemment impliqué dans les dystrophies rétiniennes. Or, dans 25% des cas, aucune ou une seule mutation n’a pu être identifiée, laissant ainsi un grand contingent de patients non résolus. Ceci s’explique d’une part car le gène ABCA4 n’est peut-être pas celui en cause chez le patient, mais d’autre part car les méthodes classiques de recherche des mutations ne couvrent pas toutes les régions des gènes, et notamment les introns.
Or on sait maintenant que ces mutations introniques profondes sont assez fréquentes. Il est donc important de développer des méthodes d’analyse du gène ABCA4 entier, afin d’identifier les mutations manquantes et pouvoir apporter un diagnostic de certitude aux patients. Le projet a également pour objectif de tester l’effet des mutations introniques identifiées et confirmer ou non leur effet pathogène. Le laboratoire du Pr CREMERS a en effet mis au point l’utilisation de petits fragments d’ADN (appelés oligonucléotides antisens), pour corriger in vitro les mutations introniques pathogènes et restaurer ainsi un effet normal, c’est à dire la production d’une protéine fonctionnelle.
L’enjeu pour les patients est majeur, car seuls ceux pour lesquels les deux mutations auront été identifiées dans ABCA4, et donc ceux pour lesquels le diagnostic de Stargardt aura été confirmé, pourront être éligibles à de futurs essais de thérapie génique.

À Nimègue (Pays-Bas), le 16 avril 2019. Rob COLLIN, Denis CAYET, Claire-Marie DHAENENS et Frans CREMERS.
Projet en thérapie génique subventionné en 2018, Thomas Lamonerie (Inserm, Nice) et Jérôme Roger (Certo, Orsay)
Le projet porte sur l’utilisation du gène CRX, gène impliqué dans la différenciation et le développement des photorécepteurs.
Les causes de la mort des cellules photoréceptrices sont génétiquement et cliniquement hétérogènes. Le succès récent du remplacement de gène NRL chez les patients avec Amaurose congénitale de Leber (LCA) causée par des mutations RPE65 a montré d’impressionnants résultats et un réel intérêt pour élargir l’approche à d’autres maladies rétiniennes. Parmi eux, les mutations CRX pathogènes représentent des candidats pertinents pour la thérapie génique en particulier grâce à des modèles de souris récemment décrits.
Le travail publié a déchiffré les mécanismes moléculaires causant un manque de vision et également démontré par différentes approches la faisabilité de restaurer la vision dans ce modèle. De tels résultats encourageants soutiennent fortement la pertinence de thérapies utilisant le virus adéno-associé (AAV) pour certaines classes de rétinopathies. En plus d’être un excellent modèle pré-clinique, ces souris pourraient également aider à jeter plus de lumières sur les mécanismes régulant l’homéostasie (maintien de équilibre naturel) des photorécepteurs.
L’équipe propose de tester la thérapie génique sur ce modèle de souris en exprimant le type sauvage du gène CRX humain. L’approche pour atteindre cet objectif est de définir les meilleures variantes d’AAV appropriées pour restaurer la réponse des photorécepteurs.
Le projet proposé est important car d’une part, la stratégie thérapeutique pourrait être utilisée pour traiter des patients atteints de rétinopathies associées au CRX ; et d’autre part, grâce à l’identification des facteurs impliqués dans l’homéostasie des photorécepteurs, ce projet aura également un impact sur l’accélération du développement de thérapies ciblant des maladies liées à la dégénérescence des photorécepteurs dont la maladie de Stargardt.
Projet en pharmacologie subventionné en 2016 et en 2017, Philippe Brabet (Inserm, Montpellier)
L’objet de la recherche porte sur le moyen de prévenir la progression de la maladie de Stargardt par l’utilisation de molécules qui ciblent à la fois la cause et ses conséquences.
Rappel sur la définition de la pharmacologie « La pharmacologie est une discipline scientifique du vivant qui étudie les mécanismes d’interaction entre une substance active et l’organisme dans lequel elle évolue, de façon à pouvoir ensuite utiliser ces résultats à des fins thérapeutiques, comme l’élaboration d’un médicament (principalement) ou son amélioration. »
L’approche pharmacologique qui consiste à traiter avec un médicament est peut-être la plus polyvalente car elle peut intervenir tôt et s’appliquer toute la vie. Toutefois, il faut trouver des molécules qui ciblent bien les deux tissus rétiniens (photorécepteurs et épithélium pigmentaire rétinien-EPR) sans effets secondaires néfastes.
Pour l’étude pharmacologique subventionnée en 2016 et 2017, l’équipe a testé de nouvelles molécules développées, en collaboration avec deux équipes françaises en pharmacie à Montpellier (IBMM, UMR5247-CNRS-UM-ENSCM) et Clermont-Ferrand (UMR1107-Inserm), à partir d’une molécule naturelle extraite d’algues, le phloroglucinol. Celle-ci possède des propriétés reconnues anti-oxydantes et antispasmodique.
L’équipe a également démontré son efficacité pour protéger les photorécepteurs et l’EPR contre une toxine élaborée par le photorécepteur, le rétinal tout-trans, source de la lipofuscine (article scientifique soumis pour publication). L’objectif des premières modifications chimiques du phloroglucinol était d’améliorer la biodisponibilité rétinienne et la réactivité vis-à-vis du rétinal tout-trans (Biodisponibilité : Proportion de molécule thérapeutique libérée à partir de la forme pharmaceutique administrée devenant disponible pour produire l’effet biochimique attendu). Deux molécules ont montré une excellente protection contre la toxicité évaluée au cours de plusieurs tests sur des cultures cellulaires.
Ces données ont permis alors d’évaluer leurs effets protecteurs dans la rétine de souris porteuses d’un gène Abca4 muté, modèle de la pathologie :
- D’une part, une batterie de tests morphologiques et fonctionnels a été effectuée
- D’autre part, ont été étudiés le mécanisme d’action des molécules, leur devenir, comprendre les mécanismes de protection mis en jeu en mesurant leur impact sur les défenses enzymatiques et non enzymatiques dans la cellule et si nécessaire faire évoluer leur structure chimique pour sélectionner les molécules les plus puissantes et améliorer leur biodisponibilité avec le moins d’effet secondaire.
Les résultats actuels orientent l’équipe vers l’élaboration de composés « lipophénoliques » ayant des effets protecteurs évalués chez une souris modèle de la maladie de Stargardt et n’ayant pas d’effets indésirables sur la rétine. Cet effet protecteur est lié à des mécanismes moléculaires reposant d’une part sur une réduction de la quantité de rétinal dans la rétine et d’autre part sur l’activation des défenses cellulaires impliquées dans la protection des photorécepteurs et de l’EPR.
Des études sur modèle animal se poursuivent pour optimiser l’efficacité du composé et minimiser sa toxicité et évaluer le mode d’administration pour un traitement à long terme (orale, local). Pour cela, une stratégie de valorisation de ces résultats a été mise en place par la SATT-AXLR, une société d’accélération du transfert de technologies qui permet à l’équipe de recherche de développer en parallèle un programme de maturation et un transfert à des partenaires industriels.
L’objectif final est de sélectionner des molécules qui pourront être efficaces seules ou en combinaison dans le traitement et/ou la prévention des dystrophies maculaires chez le sujet jeune et au cours du vieillissement.
Projet en thérapie génique subventionné en 2015, Achille François (Inserm, Nantes)
Rappel sur le principe de la thérapie génique (Source « Œil et génétique » Masson 2009)
« Le principe de la thérapie génique consiste à utiliser l’ADN comme un médicament. Introduit au sein du noyau de cellules particulières, ce morceau d’ADN, dénommé «transgène», est capable de fabriquer, en utilisant la machinerie cellulaire endogène, » (de l’organisme) « des protéines thérapeutiques ».
Pour cette étude portant sur la thérapie génique initiée par Achille François (Inserm UMR 1089, Université de Nantes) l’équipe de recherche a testé de nouveaux vecteurs viraux recombinants. Ces vecteurs viraux exploitent la capacité du virus d’origine, l’AAV, à faire pénétrer son ADN dans le noyau des cellules mais les gènes viraux ont été remplacés par le gène ABCA4 sain. Les vecteurs viraux testés ont été développés pour optimiser l’expression du transgène au niveau des photorécepteurs et permettre la production de protéine ABCA4 fonctionnelle dans ces cellules. Ils sont donc particulièrement adaptés au traitement de la maladie de Stargardt.
Des tests ont d’abord été réalisés sur des cellules in vitro (c’est-à-dire dans des boîtes de culture en laboratoire, hors organismes vivants). Les résultats encourageants ont amené l’équipe à continuer les recherches sur des rongeurs, ce qui a permis de montrer une expression du transgène ABCA4 dans la rétine après administration des vecteurs. Ils entrevoient ainsi l’espoir, d’ici quelques années, de développer des essais chez l’homme.